Mucopolysaccharidosis is the common name for a number of rare diseases that are genetic in nature. Pathology develops due to a lack of certain enzymes in the body that help break down fats and carbohydrates into simple molecules. This article discusses type 1 mucopolysaccharidosis - Hurler's syndrome.
Causes
The disease is hereditary in an autosomal recessive manner. It develops due to abnormalities in the exchange of mucopolysaccharides.
Pathogenesis
Mucopolysaccharidosis refers to the so-called lysosomal storage diseases. As a result of a deficiency of lysosomal enzymes, the catabolism of glycosaminoglycans is impeded. They accumulate in tissues and organs, disrupting the body and its systems. First of all, the skeleton is damaged and physical development is retarded.
External signs and symptoms of the disease
Symptoms of the disease are manifested in the form of defects in bone, connective, and cartilage tissues. The main symptom is stunted growth. This symptom can be detected earlier than the others, usually by the end of the first year of life it becomes clear that the child is stunted.
Mucopolysaccharidosis can also be assumed by seeing gross facial features. Patients have a large tongue, hypertelosirm (too large a distance between the paired organs, in this case between the eyes), deformed auricles, forehead overhangs, teeth are crooked.
Symptoms of mucopolysaccharidosis include chest deformities, severe kyphosis of the thoracolumbar spine. When conducting x-rays, you can detect premature ossification of the occipital-parietal suture without disturbing the ossification nuclei.
In most cases, the disease accompanies the limitation of joint mobility, abdominal hernias, hepatosplenomegaly (enlarged liver and spleen due to pathological processes resulting from the disease).
From the side of neurology, motor inhibitions and muscle hypotension are noted. Also, with mucopolysaccharidosis, hearing loss and a decrease in intelligence occur, up to severe dementia. Due to progressive systemic lesions of the skeleton, internal organs are also affected to varying degrees.
Types of Mucopolysaccharidosis
There are several types of diseases that differ in the severity of bone changes and mental disorders:
- I - Gurler's syndrome.
- II - Gunther (Hunter) syndrome.
- III - Sanfilippo syndrome.
- IV - Morkio syndrome.
- VI - Maroto-Lamy syndrome.
- VII - Sly's syndrome.
The division in medical practices of different countries may vary. Type V usually produces Cheye syndrome. In the American community of patients with mucopolysaridosis, the severity of symptoms of the first type is divided according to the severity and three phenotypes are distinguished: Gurler's syndrome, Scheye syndrome and between them Hurler-Scheieu syndrome (of which Gurler is the most severe, Scheieu is the lightest).
Hurler's Syndrome
This form is more common than others and has been described before other syndromes. In addition, the clinical picture is the most striking and typical of all types of mucopolysaccharidosis.
Hurler's syndrome develops as a result of autosomal recessive inheritance. This type of disease is characterized by very rapid progress. Despite the fact that mucopolysaccharidosis of the first type is similar to the second (Gunther, or Hunter), this is a more complex disease. For the first time, this form was described in 1919 by Gertrude Hurler (therefore, the correct name is Hurler's syndrome, not Hurler's). The frequency of occurrence is one case per 20-25 thousand people, and in most cases the parents of a sick child are in close relationship. Therefore, if a diagnosis of "Hurler’s syndrome" is made, the causes must be sought at the genetic level. Symptoms appear almost immediately after birth, and by a year or two the clinical picture is already fully expressed.
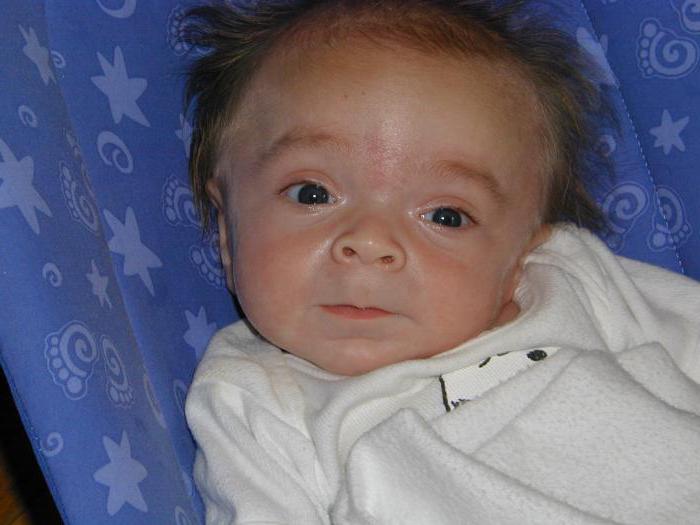
Hurler's syndrome is a classic manifestation of the disease. As the disease develops, growth slows down, visible clouding of the cornea is noted, the vessels of the nose are full of blood. With this form of the disease, an X-ray examination reveals the expansion of the Turkish saddle, shortening and expansion of long bones, hypoplasia and sharpness of the vertebrae of the lumbar region (the so-called fish vertebrae), deformations of the spinal column (patients suffer from kyphosis and lordosis of the thoracolumbar region of the spine). Pathologies of the cardiovascular system begin - the coronary arteries become clogged, valves, myocardium, endocardium change, the heart grows in size.
Hydrocephalus is noted, caused by deposits of mucopolysaccharides in the meninges. The foci of demyelination are determined. Mucopolysaccharides are also deposited in the liver, spleen, and epithelium of the renal tubules; retina, sclera, cornea; nerve cells, cartilage.
Children are already born with a characteristic appearance - they have very peculiar, rough facial features, which is why another name for mucopolysaccharoidosis is gargoyleism (from the word “gargoyle” - a fantastic figure with unusual facial features), including the so-called Hurler’s syndrome. Photos, which depict patients, clearly illustrate the bizarre distortion of the facial features of the child. The skull of these children has been changed - it takes the form of a keel of a boat, there is the so-called scaphocephaly, sunken nose, thick lips, large tongue, steep forehead, short neck and characteristic facial expression. Outwardly, it really resembles the way mythological gargoyles are portrayed.

They also have a shortened chest, lower ribs protruding, there are signs of kyphosis, the joints (especially the fingers and elbows) are inactive, there may be inguinal and umbilical hernias. Nails can take the form of watch glasses, hair becomes hard and dry, voice - low and hoarse. Hearing loss or even deafness is likely. Patients often suffer from caries, which provokes Hurler’s syndrome.
Symptoms include pathologies of the respiratory system, because of this, the child breathes with his mouth, he has adenoids, he is prone to viral infections. Over time, he develops liver and spleen problems characteristic of mucopolysaccharidoses (as a result, the abdomen is enlarged), dementia.
Growth remains dwarf. Due to improper physique and deformation of the spine, patients walk on bent legs, on tiptoe.
Gurler's syndrome has a progressive malignant nature, therefore, disability of patients occurs extremely quickly. Many do not even live to be 10 years old.
Diagnostics
The patient needs to conduct clinical, radiological, biochemical, genealogical, as well as molecular genetic studies. Diagnosis is based on the clinical manifestations of the disease, based on x-ray studies and urinalysis, which determines the activity of enzymes and the excretion of glycosaminoglycans.
Treatment of mucopolysaccharidosis
If the patient is diagnosed with Hurler’s syndrome, treatment is expected to be more symptomatic. The patient is observed comprehensively by an orthopedist, surgeon, pediatrician, otolaryngologist, neurosurgeon, ophthalmologist and neuropathologist. The patient undergoes orthopedic correction of disorders of the musculoskeletal system, hernias are removed, viral diseases, hearing disorders, otitis media, sinusitis often occurring in such patients are treated. Also under observation is the cardiovascular system.
Hormonal drugs are used that temporarily improve the patient's condition:
- glucocorticoids,
- corticotropin,
- thyroidin.
In addition, the patient is shown vitamin A, dextran 70, which also temporarily improves the patient's condition. Short-term improvement is given by transfusion of blood plasma preparations.
With Hurler's syndrome, a physiotherapeutic treatment can be prescribed to the patient: lidase electrophoresis on the affected joints, laser puncture, magnetotherapy, paraffin baths. Also, patients are recommended to engage in physical therapy, the exercises of which affect the joints and spine. Good results are often given by massage.
Since patients with Hurler's syndrome are susceptible to respiratory diseases, it is necessary to timely repair the foci of infection in the mouth and nasopharynx.
For the treatment of type 1 mucopolysaccharidosis, surgical interventions are often performed - corneal transplantation and correction of valvular heart defects and nerve injuries. In international practice, in addition to symptomatic treatment with medications, physiotherapy or surgical interventions, enzyme replacement therapy and stem cell transplantation are used.
If necessary, the patient undergoes hernia excision, removal of adenoids, antiglaucomatous operations, tracheostomy, prosthetics of the hip joints, bypass surgery for hydrocephalus, etc.
Forecast
The prognosis is unfavorable for both Hurler’s syndrome and other forms that mucopolysaccharidosis has. Gurler's syndrome is most hopeless. Skeleton changes increase every year, as a result, organs and systems are subject to more significant violations. If the child does not die from pneumonia at an early age, then by the age of 7-12 it is already physically and mentally disabled who is helpless. Units survive to adolescence.
Prevention
It is impossible to prevent this disease. But you can find it at an early stage - with prenatal diagnosis. To do this, an analysis of amniotic cells for enzyme deficiency is performed (in case of a positive result, a pregnant woman is recommended abortion).
Due to early diagnosis and timely treatment of developed compression of the spinal cord, irreversible nerve damage can be avoided. For prevention, a medical genetic consultation is mandatory.
Treatment prognosis
Despite the difficulties in treating Hurler’s syndrome, bone marrow transplantation has been used in many developed countries over the past 20 years, which significantly improves the quality of life of patients. For more than 10 years, substitution therapy drugs have been used to treat all manifestations of non-neurological mucopolysaccharidosis.